Bioinformatics for Evolutionary Biology
Topic 7: SNP calling with GATK
Accompanying material
In this tutorial we’re going to call SNPs with GATK.
The first step is again to set up directories to put our incoming files.
cd ~
mkdir log
mkdir gvcf
mkdir db
mkdir vcf
mkdir ref
mkdir bams
We also have a few programs we’re going to use. Since we will be calling them repeatedly, we’re going to save their full paths asv ariable. This will only last for the current session so if you log out you’ll have to set them up again.
gatk=/mnt/software/gatk-package-4.2.2.0-local.jar
picard=/mnt/software/picard.jar
There are 4 different samples we will be mapping today - 4 individuals [i1, i2, i8, i9] from 1 population [p1])
We’re going to have to run multiple steps on each sample, so to make this easier, we make a list of all the sample names.
You should have generated these bams last tutorial, but if you had any problems and you can copy the *bam files and their index (*.bai) to your newly generated ~/bams directory
(cp /mnt/data/bams/SalmonSim.p1.3.i[1-2,8-9].400000.sort.bam* ~/bams/).
ls bams/ | grep .sort.bam$ | sed s/.400000.sort.bam//g > samplelist.txt
Lets break this down.
ls bams <= List all the files in the bams directory
| grep .sort.bam$ <= Only keep the file names ending ($) in .sort.bam.
| sed s/.400000.sort.bam//g <= Replace the string 400000.sort.bam_ with “”, effectively leaving only the sample name.
> samplelist.txt <= Save the result in a file named samplelist.txt
The first step is to mark duplicate reads using picardtools - this is important because we wouldn’t expect to find reads that are exactly identical (the same sequence, with the same start and end positions) unless it resulted from PCR amplification. We don’t want to infer genome-wide variation with removing this pseudoreplicated data. If you were using GBS data you wouldn’t want to do this step.
#this program is sightly different than we've been working with. They are compiled with java code and contain many modules.
#we therefore need to 1) call java 2) point java to the executable 3) tell picard which module to run
#remember that you can figure out a program's usage by running it without any options except --help (i.e. java -jar $picard MarkDuplicates --help)
while read name; do
java -jar $picard MarkDuplicates \
I=~/bams/$name.400000.sort.bam O=~/bams/$name.sort.dedup.bam \
M=log/$name.duplicateinfo.txt
samtools index bams/$name.sort.dedup.bam
done < samplelist.txt
Now in the bam files duplicate reads are flagged. Take a look in the log directory, which sample has the highest number of duplicate reads?
To use GATK, we have to index our reference genome. An index is a way to allow rapid access to a very large file. For example, it can tell the program that the third chromosome starts at bit 100000, so when the program wants to access that chromosome it can jump directly there rather than scan the whole file (we’ve already done this for our bam files –> .bai) . Some index files are human readable (like .fai files) while others are not.
cp /mnt/data/fasta/SalmonReference.fasta ~/ref/ #copy the reference to our local folder
#two types of references are needed - a sequence dictionary, which you made in the RNAseq tutorial
java -jar $picard CreateSequenceDictionary \
R=~/ref/SalmonReference.fasta \
O=~/ref/SalmonReference.dict
#and a .fai file
samtools faidx ~/ref/SalmonReference.fasta #column 1 = chromsome number, c2 = length, c3 = cumulative position where contig seq begins (i.e. is not missing)
Take a look at the ref/SalmonReference.fasta.fai. How many chromosomes are there and how long is each?
The next step is to use GATK to create a GVCF file for each sample. This file summarizes support for reference or alternate alleles at all positions in the genome for each individual. It’s an intermediate file we need to use before we create our final, population-level VCF file.
This step can take a few minutes so lets first test it with a single sample to make sure it works.
#note the approach we'll use for variable assignment in our for loop
#` .. ` tells bash to take the output of this command (in this case to use as a values for "name" in the for loop)
for name in `cat ~/samplelist.txt | head -n +1 `
do
java -Xmx10g -jar $gatk HaplotypeCaller \
-R ~/ref/SalmonReference.fasta \
--native-pair-hmm-threads 2 \
-I ~/bams/$name.sort.dedup.bam \
-ERC GVCF \
-O ~/gvcf/$name.sort.dedup.g.vcf
done
Check your gvcf file to make sure it has a .idx index file. If the haplotypecaller crashes, it will produce a truncated gvcf file that will eventually crash the genotypegvcf step. Note that if you give genotypegvcf a truncated file without a idx file, it will produce an idx file itself, but it still won’t work.
We would run the HaplotypeCaller on the rest of the samples, but that will take too much time, so once you’re satisfied that your script works, you can copy the rest of the gvcf files (+ idx files) from /mnt/data/gvcf into ~/gvcf.
The next step is to import our gvcf files into a genomicsDB file. This is a compressed database representation of all the read data in our samples. It has two important features to remember:
- Each time you call GenomicsDBImport, you create a database for a single interval. This means that you can parallelize it easier, for example by calling it once per chromosome.
- The GenomicsDB file contains all the information of your GVCF files, but can’t be added to, and can’t be back transformed into a gvcf. That means if you get more samples, you can’t just add them to your genomicdDB file, you have to go back to the gvcf files.
We need to create a map file to GATK where our gvcf files are and what sample is in each. Because we use a regular naming scheme for our samples, we can create that using a bash script. This is what we’re looking for:
sample1 \t gvcf/sample1.g.vcf.gz
sample2 \t gvcf/sample2.g.vcf.gz
sample3 \t gvcf/sample3.g.vcf.gz
for i in `ls gvcf/*g.vcf | sed 's/.sort.dedup.g.vcf//g' | sed 's/gvcf\///g'`
do
echo -e "$i\tgvcf/$i.sort.dedup.g.vcf"
done > ~/biol525d.sample_map
#take a look
cat ~/biol525d.sample_map
Lets break down this loop to understand how its working
for i in ... <= This is going to take the output of the commands in the ... and loop through it line by line, putting the each line into the variable $i.
ls gvcf/”*.g.vcf” <= List all files in the gvcf directory that end with .g.vcf
| sed s/.sort.dedup.g.vcf//g <= Remove the suffix to the filename so that its only the sample name remaining.
do <= Starts the part of the script where you put the commands to be repeated.
echo -e “$i\t$gvcf/$i.sort.dedup.g.vcf” <= Print out the variable $i (which is the sample name) and then a second column with the full file name along with the soft path.
done > ~/biol525d.sample_map <= Take all the output and put it into a file name biol525d.sample_map.
Next we call GenomicsDBImport to actually create the database. This command requires a list of scaffolds so we’ll make that too.
java -Xmx10g -jar $gatk \
GenomicsDBImport \
--genomicsdb-workspace-path db/Chinook_chr1 \
--batch-size 50 \
-L chr_1 \
--sample-name-map ~/biol525d.sample_map \
--reader-threads 3
With the genomicsDB created, we’re finally ready to actually call variants and output a vcf
java -Xmx10g -jar $gatk GenotypeGVCFs \
-R ref/SalmonReference.fasta \
-V gendb://db/Chinook_chr1 \
-O vcf/Chinook_p1_i12i89_chr1.vcf.gz
Now we can actually look at the VCF file
less -S vcf/Chinook_p1_i12i89_chr1.vcf.gz
Try to find an indel. Do you see any sites with more than 1 alternate alleles?
Pick a site and figure out, whats the minor allele frequency? How many samples were genotyped?
We’ve now called variants for a single chromosome, but there are other chromosomes. In this case there are only two, but for many genomes there will be thousands of contigs. Your next challenge is to write a loop to create the genomicsdb file and then VCF for each chromosome (…check out gatk’s genomicsdb documentation… is there another way to create a db for multiple chromosomes more efficiently?)
Once you have two VCF files, one for each chromosome, you can concatenate them together to make a single VCF file. We’re going to use bcftools which is a very fast program for manipulating vcfs as well as bcfs (the binary version of a vcf).
bcftools concat \
vcf/Chinook_p1_i12i89_chr1.vcf.gz \
vcf/Chinook_p1_i12i89_chr2.vcf.gz \
-O z > vcf/full_genome.vcf.gz
You’ve done it! We have a full VCF. Tomorrow we will filter our VCF file and use it for some analyses.
In the meantime, take a look at this. We’ve ran the exact same pipeline from read data simulated to a higher coverage. Check out how important coverage is for identifying high quality SNPs.
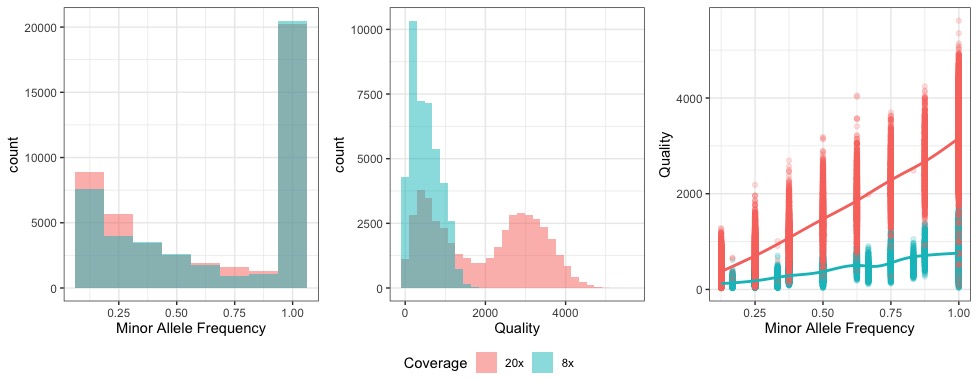
Coding Challenge
- Use command line tools to extract a list of all the samples in your VCF file, from the vcf file itself. They should be one name per line.
- Take the original vcf file produced and create a vcf of only biallelic SNPs for P1 samples.
- Use bcftools to filter your vcf file and select for sites with alternate allele frequencies > 0.01, including multi-allelic sites.